











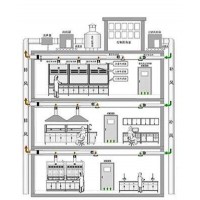


江西佰地信环境工程有限公司医疗器械净化工程-无菌洁净室工程设计的规范参照:
1、国际标准《ISO/DIS 14644》
2、洁净室厂房设计规范《GB50073-2001》
3、医疗器械包装车间洁净室厂房规范《GMP-97》
4、药品生产质量管理规范《GMP-98》
5、洁净室施工及难收规范《JGJ 71-90》
6、通风与空调工程施工及验收规范《GB 50243-2002》
7、美国联邦标准《FS209E-92》
根据相关规范要求,对无菌医疗器械生产车间、药品生产车间、医学生物学实验室、手术室等都要求建设符合相关标准的洁净室。在洁净室建设或改建时,不能依赖于最终的竣工验收来保证洁净室的质量,必须从设计及设备选型阶段就严格把关,在建设的全过程中对主要关键点严格检查、监督,在实际使用中定期监测才能保证洁净室达到设计指标和使用要求。
无菌医疗器械是任何标明“无菌”的医疗器械,生产洁净室是保证无菌医疗器械质量的基本条件,控制无菌医疗器械生产过程的环境并规范其生产,防止环境对无菌医疗器械污染,洁净室必须满足规定环境参数的要求来建设和定期监测。
江西佰地信环境工程有限公司医疗器械净化工程建设中需考虑从以下问题:
1. 医疗器械包装车间洁净室工程所需要的净化材料;
2. 医疗器械厂房洁净室及医疗器械包装车间洁净室工程的设计、安装、调试、维护等综合服务;
3. 医疗器械包装车间洁净室工程空调净化部分
温度和相对湿度
无菌医疗器械在无特殊规定时,通常要求温度在法规标准检测Standard and Testing18~28 C,湿度在 45%~65%,企业一般都可以控制在要求内。如在动态监测中发现达不到要求,可能是室内有产热大的仪器设备。
风量、换气次数、静压差
在洁净室体积确定的情况下,换气次数由该室的送风量决定,而静压差取决于房间的送风量与回风量、排风量的差值。系统总送风量、新风量、总排风量和对外压差可以通过调整风机频率转速或总阀门开启度来实现,各房间的风量和压力则可通过调整分支管路阀门开度来实现。
实际检测过程中发现,在通过调节支管风阀对某间换气次数不合格洁净室进行送风量调节时,往往会使同一洁净区其余的洁净室送风量改变,即打乱了整个洁净区的风量分配,从而使问题变得更为复杂。另外还常遇到换气次数合格而压差不合格,这种情形在二更最常见。主要原因在于护围结构气密性较差和回风口栅格不易调节。
江西佰地信环境工程有限公司
Hi,你好,欢迎来到联众商务网

